Tratamos los mecanismos fisiopatológicos del COVID 19 y otros coronavirus en el contexto de la regulación del sistema renina-angiotensina y la producción de la llamada “Tormenta de citoquinas”, procesos subyacentes a la patología (fisioetiopatogenia) de la infección por virus de ARN de la familia de los coronavirus.
Ambos sistemas están implicados en fibrosis pulmonar: una de las características fisiopatológicas de la infección por los coronavirus en general y por el COVID-19 en particular.
Fibrosis pulmonar: fisiopatología
La fibrosis pulmonar es el resultado de una lesión pulmonar y una consiguiente respuesta fibrótica que conduce al engrosamiento de las paredes alveolares y la obliteración de los espacios alveolares. Hay muchos factores tanto de origen endógeno como xenobiótico (drogas, bacterias, virus, etc.) que pueden ocasionarla.
Las principales características histológicas del pulmón fibrótico son: (Imagen)
- Daño epitelial persistente y no reparado.
- Acumulación de miofibroblastos.
- Aumento de la deposición de colágeno.

La fibrosis ocurre de manera ubicua en todo el cuerpo como una respuesta patológica a la lesión tisular crónica y es esencialmente una persistencia de la respuesta normal de curación de la herida. Se caracteriza por la inflamación crónica y la persistencia de miofibroblastos, lo que finalmente resulta en una acumulación excesiva de matriz extracelular y la destrucción de la arquitectura del tejido normal. La fibrogénesis es probablemente provocada por la inflamación, ya sea que la inflamación conduzca o no a la reparación del tejido o a la fibrosis, dependiendo del equilibrio entre la síntesis y degradación de la matriz extracelular.
En respuesta al daño tisular crónico, las células productoras de ECM, los fibroblastos, se someten a un proceso de activación caracterizado por la proliferación y diferenciación en miofibroblastos, que son los principales efectores celulares de la fibrosis1. Estas células depositan grandes cantidades de proteínas ECM y expresan la proteína contráctil α-actina del músculo liso (α-SMA), que contribuye a la disminución del cumplimiento tisular asociado con la fibrosis. Esta activación está regulada por varios factores solubles, incluidas las citoquinas, los factores de crecimiento y los productos del estrés oxidativo (2). Aunque varias moléculas están involucradas en este proceso, el factor de crecimiento transformante beta 1 (TGF-β1) desempeña un papel fundamental en la activación y el mantenimiento de la fibrogénesis (3) .
Una gran cantidad de evidencia demuestra que se requieren receptores de angiotensina II y angiotensina para la patogenia de la fibrosis pulmonar.
El sistema renina-angiotensina
La angiotensina tiene una serie de efectos profibróticos en células parenquimatosas pulmonares, que incluyen (4)
- Inducción de factores de crecimiento para células mesenquimales.
- Depósito de moléculas de la matriz extracelular.
- Aumento de producción de citoquinas.
- Aumento de la motilidad de los fibroblastos de pulmón.
- Proapoptosis de células epiteliales pulmonares.
La Angiotensina se sintetiza por el sistema local (es decir, completamente dentro del tejido pulmonar) después de una lesión pulmonar por una variedad de agentes de origen xenobiótico y endógeno.
El sistema de angiotensina consiste en el angiotensinógeno (AGT), una aspartil proteasa como la renina o la catepsina D, la enzima convertidora de angiotensina (ACE), la angiotensina II (ANGII) y los receptores de angiotensina II: tipo 1 y tipo 2 (AT1, AT2). Recientemente se ha descubierto un eje contra regulador compuesto por ACE-2, su producto , la angiotensina 1-7 (ANG1-7) y el receptor para la ANG1-7: mas (5,6)
Existe evidencia significativa in vivo que sugiere que el sistema de angiotensina está involucrado en la fibrosis pulmonar. La evidencia incluye:
- Estudios genéticos: polimorfismos genéticos del sistema ANG en pacientes con fibrosis pulmonar.
- Activación de genes del sistema ANG y sus productos proteicos en muestras de biopsia pulmonar de pacientes con fibrosis pulmonar.
- Bloqueo de la síntesis proteica de ANGII o su función: la aplicación de inhibidores de ECA ha demostrado que atenúa la fibrosis pulmonar experimental en diversos modelos (7-9).
El angiotensinógeno y la Ang II están aumentados en pacientes con fibrosis pulmonar, mostrando que la Ang II es capaz de inducir la proliferación de fibroblastos de pulmón y la producción de matriz extracelular (10-13).
Efectos antifibróticos de AT2 y del Eje ACE2 / Ang- (1-7) / Mas
Actualmente gran parte de la atención de la investigación con respecto a la angiotensina se ha dedicado a explorar la función del receptor AT2 y el receptor Mas, que, junto con ACE2 y la Angiotensina 1-7, funcionarían como el brazo protector del RAS. La reciente disponibilidad de agonistas específicos de AT2 ha proporcionado un mayor impulso en esta área, con una serie de fármacos actualmente en desarrollo preclínico o clínico (14-18).
Se ha demostrado que la estimulación del eje ACE2 / Ang- (1-7) / Mas protege contra la fibrosis pulmonar inducida por bleomicina, la lesión pulmonar inducida por aspiración y el síndrome respiratorio agudo severo (SRAS) inducido por coronavirus (19) (Figura).
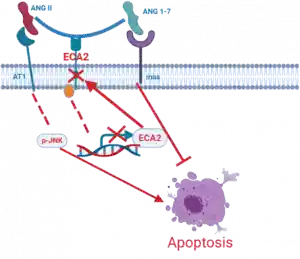
Efecto de ANG II sobre células parenquimáticas pulmonares
- La ANGII es mitogénico para los fibroblastos de pulmón humano a través de los receptores AT1 y AT2
- Los receptores AT1 y AT2 median la señalización de laANGII en el ciclo celular de fibroblastos y la migración a través de la fosforilación de Proteínas Kinasas activadas por mitógeno p38 y p42 / 44 (20,21)(Figura 1).
- En fibroblastos de pulmón humano promueve la síntesis de matriz extracelular (ECM) (22).
- Activa la producción de TGF-β (Figura 1) , involucrada en la regulación de la homeostasis y la reparación de los tejidos, la respuestas inmune e inflamatoria, la deposición de ECM, la diferenciación celular y el crecimiento (23,24).
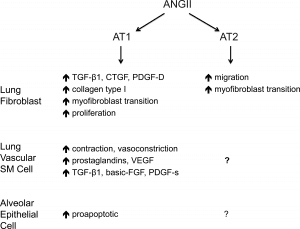
Inmunopatogénesis por Coronavirus

El coronavirus causante del síndrome respiratorio agudo severo (SARS-CoV) es el agente causante del SARS, caracterizado con neumonía atípica y fibrosis pulmonar. La enzima convertidora de angiotensina 2 (ACE2) es el principal receptor de SARS-CoV.
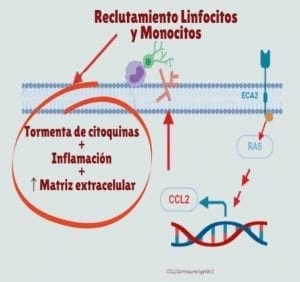
La desregulación de citoquinas inflamatorias (Cytokine storm) y moléculas de adhesión puede estar involucrada en la lesión pulmonar que causa el síndrome de estrés respiratorio agudo. Altos niveles de citoquinas proinflamatorias como la interleuquina-6, el factor de crecimiento transformante (TGF) y el factor de necrosis tumoral alfa (TNF-α) se detectan en los sueros y células ACE2 de pacientes con SARS (29,30).
Es posible que la alta tasa de mortalidad del SARS resulte de una respuesta inmune severa causada por citocinas y quimiocinas. Eventos similares son evidenciados en la literatura más reciente sobre el coronavirus COVID-19 (32).
Los mecanismos identificados de inmunopatogénesis por coronavirus incluyen:
- La CCL2 [quimiocina (motivo C-C) ligando 2) es una quimiocina CC que atrae monocitos, linfocitos T de memoria y basófilos. CCL2 y su receptor CCR2 están involucrados en reacciones inflamatorias, que incluyen la migración de monocitos / macrófagos, la polarización de células Th2, y la producción de TGF y procolágeno en fibroblastos (31). Su expresión resulta aumentada por infección con el coronavirus SARS (33) (Figura).

- Al igual que el coronavirus del síndrome respiratorio agudo severo (SARS-CoV), el coronavirus humano (HCoV)-NL63 emplea la enzima convertidora de angiotensina 2 (ACE2) como receptor para la entrada celular. La infección por SARSCoV y por (HCoV)-NL63 causa una regulación negativa de los niveles de expresión de ACE2 celular, sugiriéndose que está involucrada en la gravedad de la enfermedad (34) (Figura).
Implicaciones de este sistema regulador y la vitamina D: Vitamina D como Inmunomodulador y antifibrótico

Los estudios de intervención muestran que el bloqueo del RAS puede mejorar la fibrosis pulmonar. La hipótesis con mayor evidencia apunta a que la producción de Ang II aumenta después de una lesión pulmonar, como resultado del aumento de la expresión de angiotensinógeno en células epiteliales pulmonares dañadas y miofibroblastos activados, para estimular la fibrogénesis (22).
El sistema renina-angiotensina está sometido a una regulación vitamina D dependiente, motivo por el cual una deficiencia de esta vitamina se asocia tanto a fibrosis pulmonar como al desarrollo de síndrome respiratorio agudo.
La deficiencia de vitamina D (VDD) está estrechamente asociada con enfermedades pulmonares, como asma, fibrosis quística intersticial, enfermedad pulmonar, enfermedad pulmonar obstructiva crónica (EPOC) e infecciones respiratorias (25), y puede proteger frente a infecciones del tracto respiratorio (38).
La ingesta de Vitamina D:
- Disminuye la exacerbación de la EPOC y mejora el volumen espiratorio forzado en un segundo (VEF1) en pacientes con EPOC grave y muy grave (26).
- La vitamina D y su receptor juegan un papel vital en la fibrosis de pulmón (27) a través de una regulación negativa del:
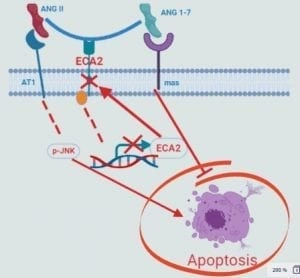
- Factor nuclear kappaB (NF-κB) , la β-catenina , que, junto con la inmunomodulación controlan el “Cytokine Storm” (28-35) (Figura 1).
- Sistema renina-angiotensina (RAS) afectado por infección de coronavirus que puede dar lugar a inflamación, la apoptosis de células pulmonares y el depósito de matriz extracelular, ocasionando fibrosis (36,37) (Figura 2).
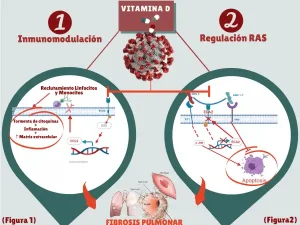
Autor: Ruth Matute, Directora científica de Equisalud.
Bibiografía
1. Hinz, B., Phan, S. H., Thannickal, V. J., Galli, A., Bochaton-Piallat, M. L., & Gabbiani, G. (2007). The myofibroblast: one function, multiple origins. The American journal of pathology, 170(6), 1807-1816..
2. P01owell, D. W., Mifflin, R. C., Valentich, J. D., Crowe, S. E., Saada, J. I., & West, A. B. (1999). Myofibroblasts. I. Paracrine cells important in health and disease. American Journal of Physiology-Cell Physiology, 277(1), C1-C19.
3.Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-β signaling in fibrosis. Growth Factors. 2011;29:196–202.
4. Budinger, GR Scott. “Angiotensin II and pulmonary fibrosis, a new twist on an old story.” American Journal of Physiology-Lung Cellular and Molecular Physiology3 (2011): L267-L268.
5. Burrell LM, Johnston CI, Tikellis C, Cooper ME. ACE2, a new regulator of the renin-angiotensinsystem. Trends. Endocrinol. Metab. 2004; 15:166–169. [PubMed: 15109615]
6. Li X, Shu R, Filippatos G, Uhal BD. Apoptosis in lung injury and remodeling. J. Appl. Physiol.
7. Li X, Zhuang J, Rayford H, Zhang H, Shu R, Uhal BD. Attenuation of bleomycin-inducedpulmonary fibrosis by intratracheal administration of antisense oligonucleotides against angiotensinogen mRNA. Pharm. Des. 2007; 13:1257–1268. [PubMed: 17504234]
8. Wang R, Ibarra-Sunga O, Verlinski L, Pick R, Uhal BD. Abrogation of bleomycin-inducedepithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am. J. Physiol. LungCell Mol. Physiol. 2000; 279:143–151.
9. Molteni A, Wolfe LF, Ward WF, Ts’ao CH, Molteni LB, Veno P, Fish BL, Taylor JM, QuintanillaN, Herndon B, Moulder JE. Effect of an angiotensin II receptor blocker and two angiotensinconverting enzyme inhibitors on transforming growth factor-beta (TGF-beta) and alpha-actomyosin(alpha SMA), important mediators of radiation-induced pneumopathy and lung fibrosis. Curr.
10. Des. 2007; 13:1307–1316. [PubMed: 17506716]2004; 97:1535–1542. [PubMed: 15358756]
11. Li, X. et al. Extravascular sources of lung angiotensin peptide synthesis in idiopathic pulmonary fibrosis. Am J Physiol Lung CellMol Physiol 291, L887–895 (2006).
12. Konigshoff, M. et al. The angiotensin II receptor 2 is expressed and mediates angiotensin II signaling in lung fibrosis. Am J RespirCell Mol Biol 37, 640–650 (2007).
13. Uhal, B. D., Li, X., Piasecki, C. C. & Molina-Molina, M. Angiotensin signalling in pulmonary fibrosis. Int J Biochem Cell Biol 44,465–468 (2012
14. Bader M, Santos RA, Unger T, Steckelings UM. New therapeutic pathways in the RAS. J Renin Angiotensin Aldosterone Syst. 2012;13:505–8.
15. Rompe F, Artuc M, Hallberg A, Alterman M, Stroder K, Thone-Reineke C, et al. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappaB. Hypertension. 2010;55:924–31.
16. Meffert S, Stoll M, Steckelings UM, Bottari SP, Unger T. The angiotensin II AT2 receptor inhibits proliferation and promotes differentiation in PC12W cells. Mol Cell Endocrinol. 1996;122:59–67.
17. Sumners C, Horiuchi M, Widdop RE, McCarthy C, Unger T, Steckelings UM. Protective arms of the renin-angiotensin-system in neurological disease. Clin Exp Pharmacol Physiol. 2013;40:580–8.
18. Santos RAS, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216:R1–R17.
19. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–9.
20. Königshoff M, Wilhelm A, Jahn A, Sedding D, Amarie OA, Eul B, Seeger W, Fink L, Günther A, Eickelberg O, Rose F. The angiotensin II receptor 2 is expressed and mediates angiotensin IIsignaling in lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2007; 37:640–650. [PubMed: 17630322].
21. Marshall RP, McAnulty RJ, Laurent GJ. Angiotensin II is mitogenic for human lung fibroblasts viaactivation of the type 1 receptor. Am. J. Respir. Crit. Care Med. 2000; 161:1999-04. [PubMed:10852780]
22. Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal. Immunol. 2009; 2:103–121. [PubMed: 19129758]
23. Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–7.4
24. Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83–92.
25. Finklea, J. D., Grossmann, R. E. & Tangpricha, V. Vitamin D and chronic lung disease: a review of molecular mechanisms and clinical studies. Adv Nutr 2, 244–253 (2011).
26. Zendedel, A. et al. Effects of Vitamin D Intake on FEV1 and COPD Exacerbation: A Randomized Clinical Trial Study. Glob J HealthSci 7, 243–248 (2015).
27. Zhang, Z. et al. Preventive effects of vitamin D treatment on bleomycin-induced pulmonary fibrosis. Sci Rep 5, 17638 (2015).
28. Mirković, K. & de Borst, M. H. Beyond the RAAS: dissecting the antifibrotic effects of vitamin D analogues. Lab Invest 92,66–1669 (2012).
29. He, L., Y. Ding, Q. Zhang, X. Che, Y. He, H. Shen, H. Wang, Z. Li, L. Zhao,J. Geng, Y. Deng, L. Yang, J. Li, J. Cai, L. Qiu, K. Wen, X. Xu, and S. Jiang. Expression of elevated levels of pro-inflammatory cytokines in SARSCoV-infected ACE2_ cells in SARS patients: relation to the acute lunginjury and pathogenesis of SARS. J. Pathol. 210:288–297.
30. A. Leeson, A. Andonov, Y. Li, N. Bastien, J. Cao, C. Osiowy, F. Dobie,.Cutts, M. Ballantine, and X. Li. 2003. Activation of AP-1 signal transductionpathway by SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 311:870–876.
31. Gharaee-Kermani, M., E. M. Denholm, and S. H. Phan. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J. Biol. Chem. 271:17779–1778
32. In the eye of the COVID-19 cytokine stormNatalie Vaninov .Nature Reviews Immunology (2020).
33. Chen, I-Yin, et al. “Upregulation of the chemokine (CC motif) ligand 2 via a severe acute respiratory syndrome coronavirus spike-ACE2 signaling pathway.” Journal of virology 84.15 (2010): 7703-7712.
34. Dijkman, Ronald, et al. “Replication-dependent downregulation of cellular angiotensin-converting enzyme 2 protein expression by human coronavirus NL63.” Journal of general virology 93.9 (2012): 1924-1929.
35. Skrobot, Agnieszka, Urszula Demkow, and Małgorzata Wachowska. “Immunomodulatory role of vitamin D: a review.” Current Trends in Immunity and Respiratory Infections. Springer, Cham, 2018. 13-23.
36. Shi, Yongyan, et al. “Chronic vitamin D deficiency induces lung fibrosis through activation of the renin-angiotensin system.” Scientific reports 7.1 (2017): 1-10.
37. Dancer, Rachel CA, et al. “Vitamin D deficiency contributes directly to the acute respiratory distress syndrome (ARDS).” Thorax 70.7 (2015): 617-624. Soc. Esp. Dolor vol.23 no.4 Madrid jul./ago. 20162-BMJ 2017;356:i6583.